crispyx Tutorial
Genome-wide CRISPR screens routinely produce datasets with hundreds of thousands of cells and tens of thousands of genes. Standard single-cell toolkits (Scanpy, Pertpy) load the entire count matrix into memory, which can require 30–100+ GB of RAM. crispyx streams data directly from on-disk AnnData .h5ad files so that QC, normalisation, pseudo-bulk aggregation, and differential expression all run without materialising the full matrix.
This tutorial walks through a complete crispyx workflow on the included Adamson subset dataset, covering:
Data loading and inspection
Data preparation utilities
Quality control
Streaming normalisation
Dimension reduction (PCA, KNN, UMAP)
Pseudo-bulk aggregation
Differential expression (Wilcoxon and NB-GLM)
LFC shrinkage
Plotting
For the full API reference, see the documentation.
Setup
Install the project in editable mode (with the optional test extras) to make the package importable:
pip install -e .[test]
1. Load the dataset
This tutorial uses the included Adamson subset stored at data/Adamson_subset.h5ad. cx.read_h5ad_ondisk returns a read-only wrapper — the expression matrix stays on disk.
[1]:
import sys
from pathlib import Path
import pandas as pd
ROOT = Path('..').resolve()
sys.path.insert(0, str(ROOT))
sys.path.insert(0, str(ROOT / 'src'))
DATA_PATH = ROOT / 'data' / 'Adamson_subset.h5ad'
OUTPUT_DIR = ROOT / 'docs' / 'tutorial_outputs'
OUTPUT_DIR.mkdir(parents=True, exist_ok=True)
import crispyx as cx
if not DATA_PATH.exists():
raise FileNotFoundError(f'Expected dataset at {DATA_PATH}')
Inspect metadata
The obs and var accessors expose head and load helpers so you can preview metadata without materialising the entire object.
[2]:
adata_ro = cx.read_h5ad_ondisk(DATA_PATH)
adata_ro
AnnData object with n_obs × n_vars = 1716 × 11630 backed at '/Users/dujinhong/Library/CloudStorage/OneDrive-TheUniversityOfHongKong/Streamlining-CRISPR-Screen-Analysis/Streamlining-CRISPR-Screen-Analysis/data/Adamson_subset.h5ad'
obs: 'perturbation', 'n_genes', 'dataset', 'celltype'
var: 'Ensembl_ID'
First obs rows:
perturbation n_genes dataset celltype
Cell_barcodes
AAACATACAAGATG-1 control 2914 Adamson K562
AAACATTGCATTGG-1 control 3999 Adamson K562
AAACGGCTAATGCC-1 AMIGO3 3947 Adamson K562
AAAGACGAACTCAG-1 control 2713 Adamson K562
AAAGAGACTCGCCT-1 control 3678 Adamson K562
First var rows:
Ensembl_ID
Gene_symbol
LINC00115 ENSG00000225880
NOC2L ENSG00000188976
KLHL17 ENSG00000187961
HES4 ENSG00000188290
ISG15 ENSG00000187608
[2]:
AnnData(path=/Users/dujinhong/Library/CloudStorage/OneDrive-TheUniversityOfHongKong/Streamlining-CRISPR-Screen-Analysis/Streamlining-CRISPR-Screen-Analysis/data/Adamson_subset.h5ad, mode='r')
[3]:
adata_ro.obs.head()
[3]:
| perturbation | n_genes | dataset | celltype | |
|---|---|---|---|---|
| Cell_barcodes | ||||
| AAACATACAAGATG-1 | control | 2914 | Adamson | K562 |
| AAACATTGCATTGG-1 | control | 3999 | Adamson | K562 |
| AAACGGCTAATGCC-1 | AMIGO3 | 3947 | Adamson | K562 |
| AAAGACGAACTCAG-1 | control | 2713 | Adamson | K562 |
| AAAGAGACTCGCCT-1 | control | 3678 | Adamson | K562 |
[4]:
adata_ro.var.head()
[4]:
| Ensembl_ID | |
|---|---|
| Gene_symbol | |
| LINC00115 | ENSG00000225880 |
| NOC2L | ENSG00000188976 |
| KLHL17 | ENSG00000187961 |
| HES4 | ENSG00000188290 |
| ISG15 | ENSG00000187608 |
2. Data Preparation Utilities
crispyx provides helpers to inspect and standardise metadata in backed h5ad files without loading the expression matrix.
[5]:
# Feature 1: load obs/var without reading X
obs = cx.load_obs(DATA_PATH)
print('obs columns:', list(obs.columns))
print('obs shape: ', obs.shape)
var = cx.load_var(DATA_PATH)
print('var columns:', list(var.columns))
print('var shape: ', var.shape)
obs columns: ['perturbation', 'n_genes', 'dataset', 'celltype']
obs shape: (1716, 4)
var columns: ['Ensembl_ID']
var shape: (11630, 1)
[6]:
# Feature 4: auto-detect perturbation and gene columns
cols = cx.infer_columns(DATA_PATH)
print('Detected columns:', cols)
Detected columns: {'perturbation_column': 'perturbation', 'gene_name_column': None}
[7]:
# Feature 3: normalise perturbation labels (inplace=False for preview)
normalised = cx.normalise_perturbation_labels(
DATA_PATH,
column='perturbation',
canonical_control='NTC',
inplace=False,
)
print(normalised.value_counts().head(10))
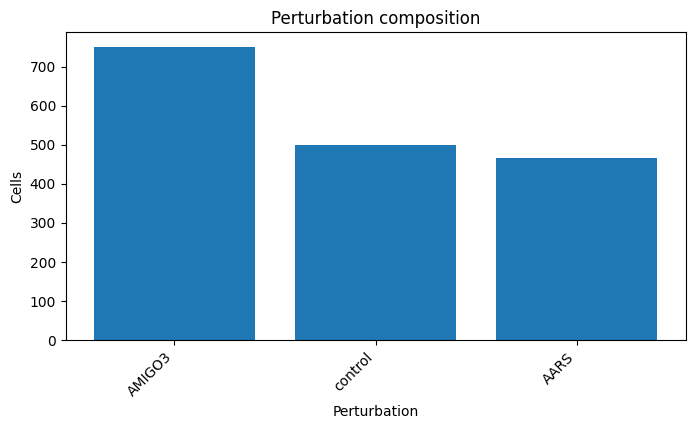
perturbation
AMIGO3 750
NTC 500
AARS 466
Name: count, dtype: int64
[8]:
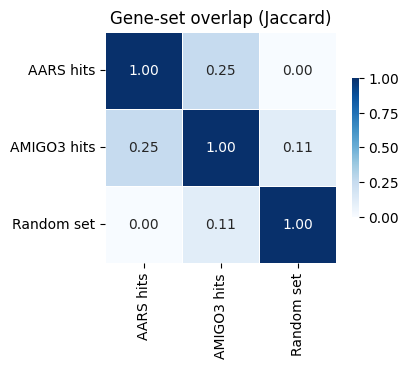
# Feature 5: overlap analysis between gene sets from different perturbations
# (Using toy sets for illustration; in practice, fill with DE-significant genes)
de_sets = {
'AARS hits': {'ATF4', 'DDIT3', 'ASNS', 'EIF2AK4', 'SLC7A5'},
'AMIGO3 hits': {'ATF4', 'DDIT3', 'CDK4', 'CDKN1A', 'EGFR'},
'Random set': {'BRCA1', 'TP53', 'KRAS', 'EGFR', 'MYC'},
}
result = cx.tl.compute_overlap(de_sets)
print('Jaccard matrix:')
print(result.jaccard_matrix.round(3))
print('\nSet sizes:', result.set_sizes.to_dict())
ax = cx.pl.overlap_heatmap(result, metric='jaccard', title='Gene-set overlap (Jaccard)')
Jaccard matrix:
AARS hits AMIGO3 hits Random set
AARS hits 1.00 0.250 0.000
AMIGO3 hits 0.25 1.000 0.111
Random set 0.00 0.111 1.000
Set sizes: {'AARS hits': 5, 'AMIGO3 hits': 5, 'Random set': 5}
3. Quality Control
The cx.pp preprocessing namespace mirrors Scanpy’s API. Each function returns a new cx.AnnData object backed by an .h5ad file on disk.
[9]:
qc_result = cx.quality_control_summary(
adata_ro,
perturbation_column='perturbation',
min_genes=5,
min_cells_per_perturbation=5,
output_dir=OUTPUT_DIR,
data_name='tutorial',
)
qc_preview = pd.DataFrame(
{
'perturbation': adata_ro.obs['perturbation'],
'qc_pass': qc_result.cell_mask,
}
)
adata_ro = qc_result.filtered
print(f"QC result: {qc_result.cell_mask.sum()} / {len(qc_result.cell_mask)} cells passed")
qc_preview.head(5)
QC result: 1716 / 1716 cells passed
[9]:
| perturbation | qc_pass | |
|---|---|---|
| Cell_barcodes | ||
| AAACATACAAGATG-1 | control | True |
| AAACATTGCATTGG-1 | control | True |
| AAACGGCTAATGCC-1 | AMIGO3 | True |
| AAAGACGAACTCAG-1 | control | True |
| AAAGAGACTCGCCT-1 | control | True |
4. Streaming Normalisation
For large datasets that would OOM during normalisation, use the streaming preprocessing function. This is the equivalent of scanpy.pp.normalize_total() + scanpy.pp.log1p() but processes data in chunks.
[10]:
# Create a normalized + log1p version of the QC-filtered data (streaming)
# This works on huge datasets without loading into memory
adata_norm = cx.pp.normalize_total_log1p(
adata_ro, # Pass the AnnData object directly (same as other cx.pp functions)
output_dir=OUTPUT_DIR,
data_name='tutorial',
target_sum=1e4, # Same as scanpy default
verbose=True,
)
# Verify the output - adata_norm is a read-only AnnData wrapper
print(f"Shape: {adata_norm.shape}")
print(f"First 3 cells, first 3 genes:\n{adata_norm.to_memory().X[:3, :3].toarray()}")
Generating preprocessed dataset (streaming, normalize+log1p): /Users/dujinhong/Library/CloudStorage/OneDrive-TheUniversityOfHongKong/Streamlining-CRISPR-Screen-Analysis/Streamlining-CRISPR-Screen-Analysis/docs/tutorial_outputs/crispyx_tutorial_normalized_log1p.h5ad
✓ Preprocessed dataset written: 1716 cells × 9238 genes
Shape: (1716, 9238)
First 3 cells, first 3 genes:
[[0.75838053 0. 0. ]
[0.49522132 0. 0. ]
[1.4766263 0. 0.5161892 ]]
5. Dimension Reduction
crispyx provides streaming PCA and KNN graph construction that works on backed (on-disk) AnnData objects.
Streaming PCA: Automatically selects the optimal method based on gene count (
sparse_covfor ≤15K genes,incrementalfor larger).KNN neighbors: Builds k-nearest neighbor graph from PCA embeddings for downstream clustering/UMAP.
Results are written directly to the h5ad file using a close-write-reopen pattern.
[11]:
# Streaming PCA on the normalized data
# Results are written directly to the h5ad file (close-write-reopen pattern)
cx.pp.pca(adata_norm, n_comps=20, show_progress=True)
# Check PCA results (data is read from the h5ad file)
print(f"PCA embeddings shape: {adata_norm.obsm['X_pca'].shape}")
print(f"Variance explained: {adata_norm.uns['pca']['variance_ratio'][:5]}") # First 5 components
# Build KNN graph from PCA embeddings (also writes to h5ad)
cx.pp.neighbors(adata_norm, n_neighbors=10, method='sklearn', show_progress=True)
print(f"KNN distances shape: {adata_norm.obsp['distances'].shape}")
print(f"Number of connections: {adata_norm.obsp['connectivities'].nnz}")
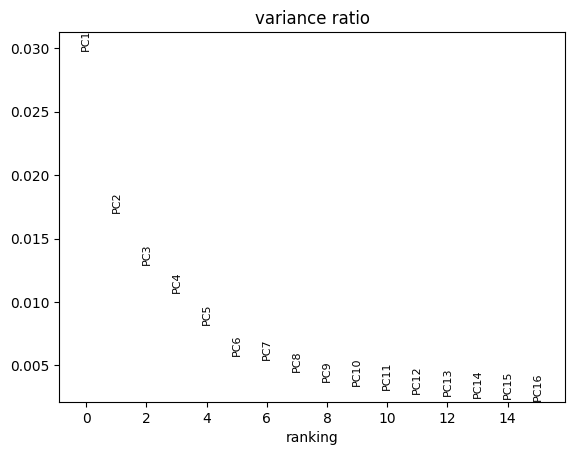
PCA embeddings shape: (1716, 20)
Variance explained: [0.02978331 0.01700597 0.01289528 0.01072035 0.00821617]
KNN distances shape: (1716, 1716)
Number of connections: 25228
PCA Visualization
The cx.pl namespace provides Scanpy-style plotting wrappers:
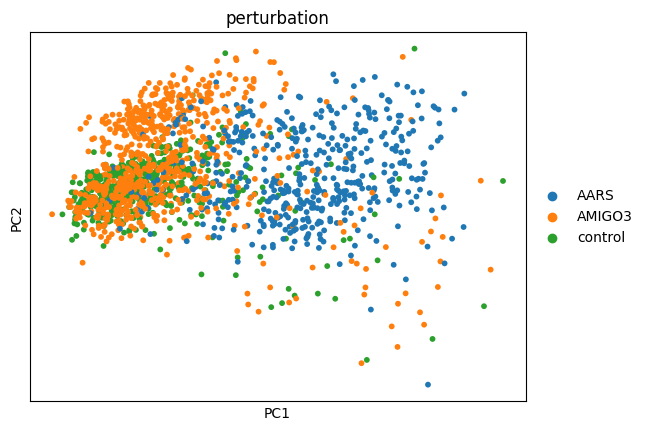
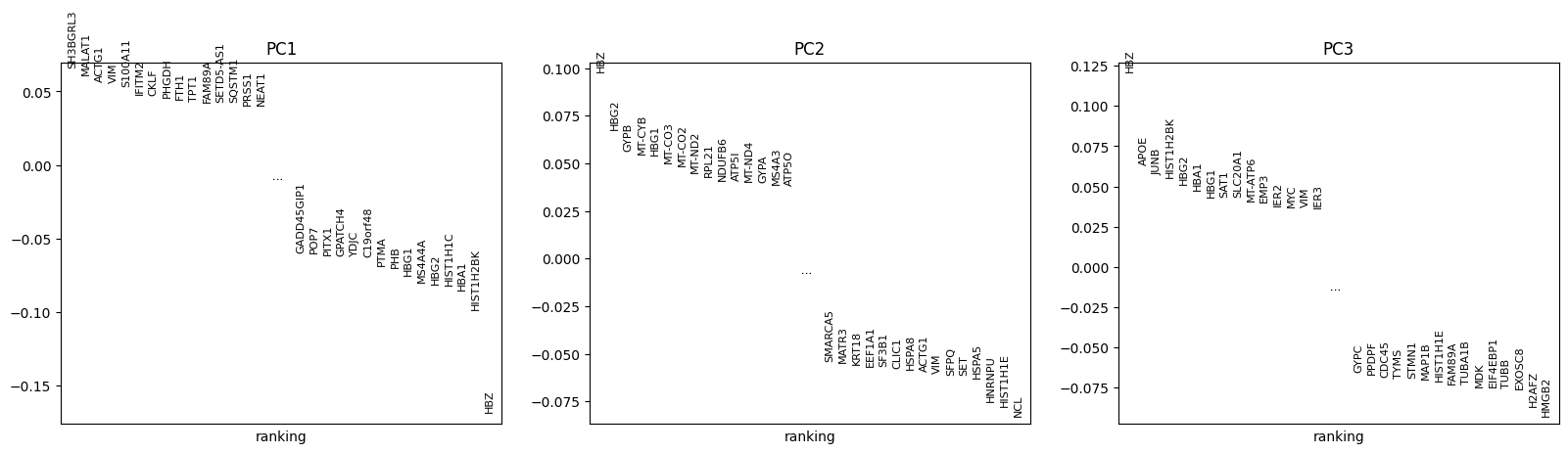
cx.pl.pca(): Scatter plot of PCA embeddingscx.pl.pca_variance_ratio(): Explained variance per componentcx.pl.pca_loadings(): Gene loadings for top components
[12]:
# Plot PCA variance explained
cx.pl.pca_variance_ratio(adata_norm, n_pcs=15)
# Plot PCA scatter colored by perturbation
cx.pl.pca(adata_norm, color='perturbation', components='1,2')
# Plot gene loadings for first 3 components
cx.pl.pca_loadings(adata_norm, components=[1, 2, 3])
UMAP Visualization
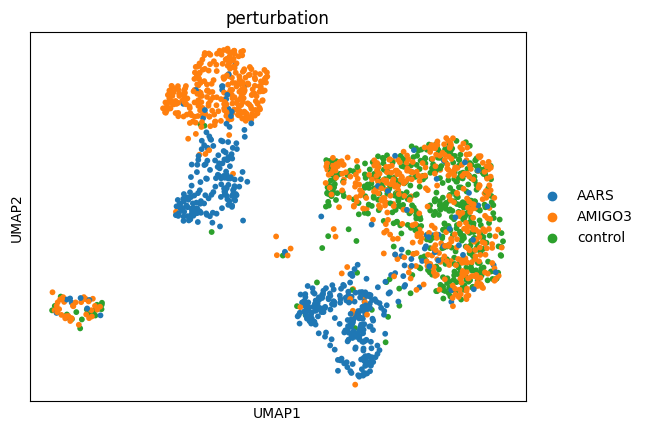
After computing PCA and building the neighbor graph, we can compute UMAP embeddings for visualization. cx.tl.umap() uses the pre-computed neighbor graph from cx.pp.neighbors(), so it only loads the neighbor matrix (not the full expression matrix) into memory.
This is memory-efficient: approximately 0.75 MB per 1000 cells for 15 neighbors.
[13]:
# Compute UMAP from neighbor graph (memory-efficient)
cx.tl.umap(adata_norm, min_dist=0.5, spread=1.0)
# Check UMAP results
print(f"UMAP embedding shape: {adata_norm.obsm['X_umap'].shape}")
# Plot UMAP colored by perturbation
cx.pl.umap(adata_norm, color='perturbation')
WARNING: .obsp["connectivities"] have not been computed using umap
UMAP embedding shape: (1716, 2)
Pseudobulk aggregation with cx.pb
Aggregate cells per perturbation on disk. The returned object can be inspected lazily, similar to the original dataset.
[14]:
adata_pb_ro = cx.pb.average_log_expression(
adata_ro,
perturbation_column='perturbation',
output_dir=OUTPUT_DIR,
data_name='tutorial',
)
print(f"Pseudobulk shape: {adata_pb_ro.shape}")
adata_pb_ro.var.head(5)
Pseudobulk shape: (2, 9238)
[14]:
| Gene_symbol |
|---|
| NOC2L |
| KLHL17 |
| HES4 |
| ISG15 |
| AGRN |
Differential expression with cx.tl
Run a Wilcoxon test that mirrors scanpy.tl.rank_genes_groups. cx.AnnData.uns entries also provide preview and load helpers.
[15]:
adata_ro = cx.tl.rank_genes_groups(
adata_norm, # Wilcoxon expects log-normalized data
perturbation_column='perturbation',
method='wilcoxon',
output_dir=OUTPUT_DIR,
data_name='tutorial',
)
# Preview a tidy DE table using plotting helper (no scanpy_format needed)
group = adata_ro.obs.load()['perturbation'].astype(str).unique()[0]
cx.pl.rank_genes_groups_df(adata_ro, group=group, n_genes=10).head()
[15]:
| names | scores | logfoldchanges | pvals | pvals_adj | pts | pts_rest | group | |
|---|---|---|---|---|---|---|---|---|
| 0 | SET | -10.899647 | -0.506634 | 1.157049e-27 | 8.419143e-24 | 0.990667 | 0.988 | AMIGO3 |
| 1 | MT-CO2 | 10.858218 | 0.402062 | 1.822720e-27 | 8.419143e-24 | 0.998667 | 0.998 | AMIGO3 |
| 2 | RPS27A | 10.551140 | 0.267033 | 5.018421e-26 | 1.022991e-22 | 1.000000 | 1.000 | AMIGO3 |
| 3 | EIF1 | 10.539785 | 0.288907 | 5.662821e-26 | 1.022991e-22 | 1.000000 | 1.000 | AMIGO3 |
| 4 | MT-CYB | 10.534347 | 0.327073 | 5.999814e-26 | 1.022991e-22 | 0.998667 | 0.998 | AMIGO3 |
Use load to materialise the complete differential expression tables for downstream analysis.
[16]:
group = adata_ro.obs.load()['perturbation'].astype(str).unique()[0]
top_df = cx.pl.rank_genes_groups_df(adata_ro, group=group, n_genes=5)
print(f"Top 5 DE genes for perturbation '{group}':")
top_df[['names', 'logfoldchanges', 'pvals_adj']]
Top 5 DE genes for perturbation 'AMIGO3':
[16]:
| names | logfoldchanges | pvals_adj | |
|---|---|---|---|
| 0 | SET | -0.506634 | 8.419143e-24 |
| 1 | MT-CO2 | 0.402062 | 8.419143e-24 |
| 2 | RPS27A | 0.267033 | 1.022991e-22 |
| 3 | EIF1 | 0.288907 | 1.022991e-22 |
| 4 | MT-CYB | 0.327073 | 1.022991e-22 |
The cx.AnnData handles close themselves when their Python objects go out of scope, so no explicit cleanup is required.
NB-GLM Differential Expression
For more sophisticated differential expression analysis, crispyx provides a negative binomial GLM method that models count data directly. This is especially useful when you need to incorporate covariates or want more accurate statistical modeling.
Note: NB-GLM operates on raw counts (not log-normalized data), so we use the QC-filtered dataset directly.
[17]:
# Run NB-GLM test on raw counts
# First, reload the QC-filtered dataset (not log-normalized)
qc_path = OUTPUT_DIR / 'crispyx_tutorial_filtered.h5ad'
adata_qc = cx.read_h5ad_ondisk(qc_path)
adata_nb = cx.tl.rank_genes_groups(
adata_qc,
perturbation_column='perturbation',
method='nb_glm',
output_dir=OUTPUT_DIR,
data_name='tutorial_nb',
)
group = adata_nb.obs.load()['perturbation'].astype(str).unique()[0]
cx.pl.rank_genes_groups_df(adata_nb, group=group, n_genes=5).head()
AnnData object with n_obs × n_vars = 1716 × 9238 backed at '/Users/dujinhong/Library/CloudStorage/OneDrive-TheUniversityOfHongKong/Streamlining-CRISPR-Screen-Analysis/Streamlining-CRISPR-Screen-Analysis/docs/tutorial_outputs/crispyx_tutorial_filtered.h5ad'
obs: 'perturbation', 'n_genes', 'dataset', 'celltype'
var: 'Ensembl_ID', 'gene_symbols'
First obs rows:
perturbation n_genes dataset celltype
Cell_barcodes
AAACATACAAGATG-1 control 2914 Adamson K562
AAACATTGCATTGG-1 control 3999 Adamson K562
AAACGGCTAATGCC-1 AMIGO3 3947 Adamson K562
AAAGACGAACTCAG-1 control 2713 Adamson K562
AAAGAGACTCGCCT-1 control 3678 Adamson K562
First var rows:
Ensembl_ID gene_symbols
Gene_symbol
NOC2L ENSG00000188976 NOC2L
KLHL17 ENSG00000187961 KLHL17
HES4 ENSG00000188290 HES4
ISG15 ENSG00000187608 ISG15
AGRN ENSG00000188157 AGRN
[17]:
| names | scores | logfoldchanges | pvals | pvals_adj | pts | pts_rest | group | |
|---|---|---|---|---|---|---|---|---|
| 0 | MT-CO2 | 10.753535 | 0.447715 | 5.703282e-27 | 5.268692e-23 | 0.998667 | 0.998 | AMIGO3 |
| 1 | HIST1H1E | -10.207953 | -0.838292 | 1.826781e-24 | 8.437901e-21 | 0.512000 | 0.738 | AMIGO3 |
| 2 | MT-CYB | 10.039458 | 0.402310 | 1.022347e-23 | 3.148146e-20 | 0.998667 | 0.998 | AMIGO3 |
| 3 | HIST1H1D | -9.473744 | -0.889809 | 2.699881e-21 | 5.309861e-18 | 0.436000 | 0.656 | AMIGO3 |
| 4 | UQCRB | 9.467219 | 0.537892 | 2.873923e-21 | 5.309861e-18 | 0.952000 | 0.918 | AMIGO3 |
LFC Shrinkage with apeGLM
Apply adaptive shrinkage to log-fold changes using the apeGLM method. This shrinks noisy/uncertain LFCs toward zero while preserving large effects, following DESeq2/PyDESeq2 best practices.
The two-step workflow (NB-GLM → shrink_lfc) gives you control over when shrinkage is applied and allows you to compare shrunk vs unshrunk results.
[18]:
# Apply LFC shrinkage to NB-GLM results
nb_result_path = adata_nb.path
shrunk = cx.tl.shrink_lfc(
nb_result_path,
prior_scale_mode='global', # Use global prior across all perturbations
)
# Compare shrunk vs raw LFC for a perturbation
import anndata
shrunk_adata = anndata.read_h5ad(nb_result_path)
print("Shrunk vs Raw LFC (first 5 genes, first perturbation):")
print(f" Shrunk LFC: {shrunk_adata.X[0, :5]}")
print(f" Raw LFC: {shrunk_adata.layers['logfoldchange_raw'][0, :5]}")
Shrunk vs Raw LFC (first 5 genes, first perturbation):
Shrunk LFC: [ 0.00091058 -0.02613842 0.0247642 0.0387339 -0.07999716]
Raw LFC: [ 0.00091058 -0.02613842 0.0247642 0.0387339 -0.07999716]
The shrunk LFC values are typically smaller in magnitude for genes with high uncertainty, while large, well-supported effects are preserved. This improves downstream ranking and visualization.
Plotting (Scanpy-style)
crispyx provides cx.pl helpers for plotting on-disk results. The examples below mirror Scanpy style while keeping the expression matrix on disk.
[19]:
# QC plots
cx.pl.qc_perturbation_counts(
data=adata_qc,
perturbation_column='perturbation',
cell_mask=qc_result.cell_mask,
)
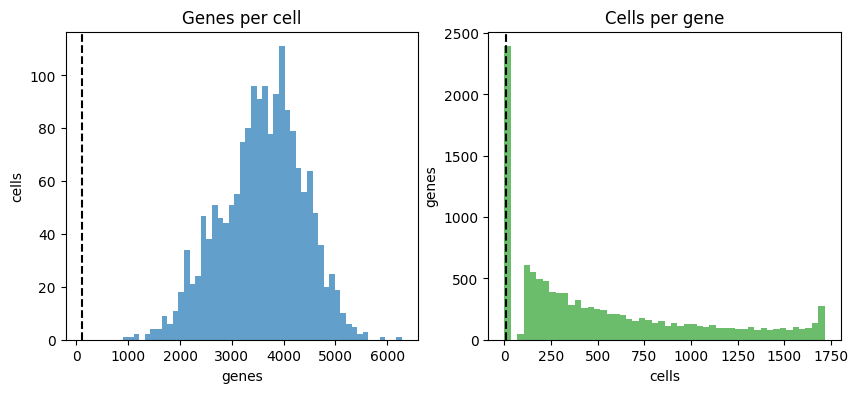
cx.pl.qc_summary(qc_result, min_genes=100, min_cells_per_gene=10)
# Rank genes groups plot
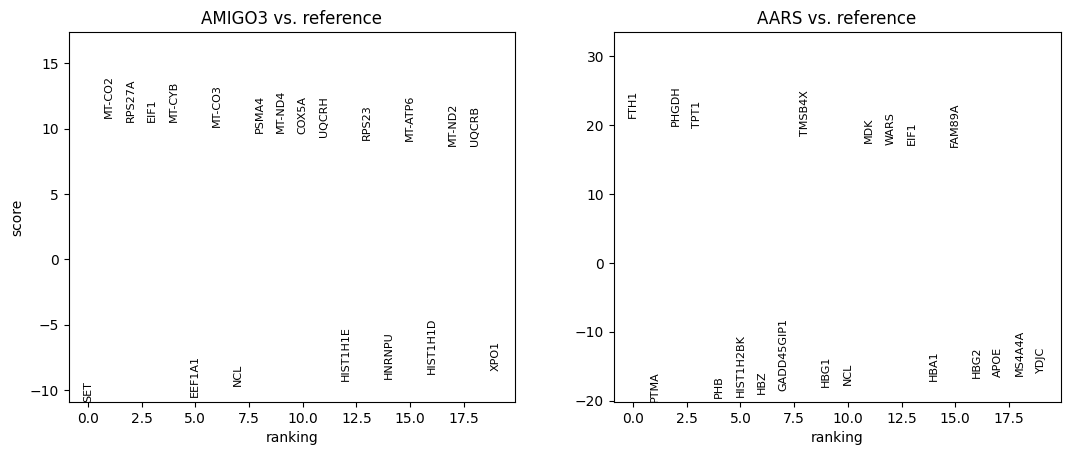
cx.pl.rank_genes_groups(adata_ro, n_genes=20, sharey=False)
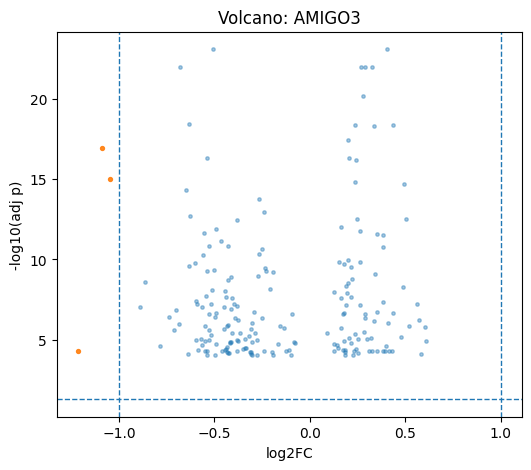
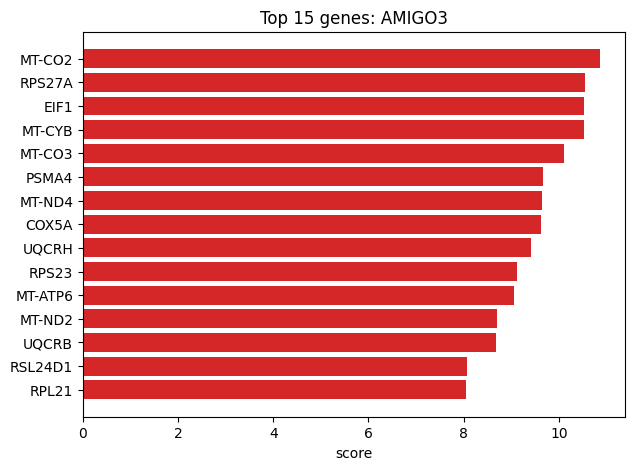
# Volcano / top genes
plot_group = adata_ro.obs.load()['perturbation'].astype(str).unique()[0]
df = cx.pl.rank_genes_groups_df(adata_ro, group=plot_group, n_genes=200)
cx.pl.volcano(de_df=df, group=plot_group)
cx.pl.top_genes_bar(de_df=df, group=plot_group, topn=15)
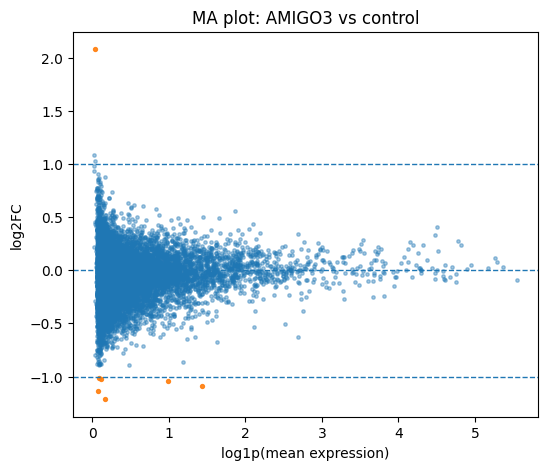
# MA plot (raw counts from QC-filtered data)
try:
control_label = adata_ro.uns['control_label'].load()
except KeyError:
control_label = 'control'
cx.pl.ma(
data=adata_qc,
de_result=adata_ro,
group=plot_group,
reference=control_label,
perturbation_column='perturbation',
mean_mode='raw',
)
[19]:
<Axes: title={'center': 'MA plot: AMIGO3 vs control'}, xlabel='log1p(mean expression)', ylabel='log2FC'>